今日の医療において、欠かせない医薬品。一つの薬が世に出るには長い時間と多額の費用がかかると言われます。
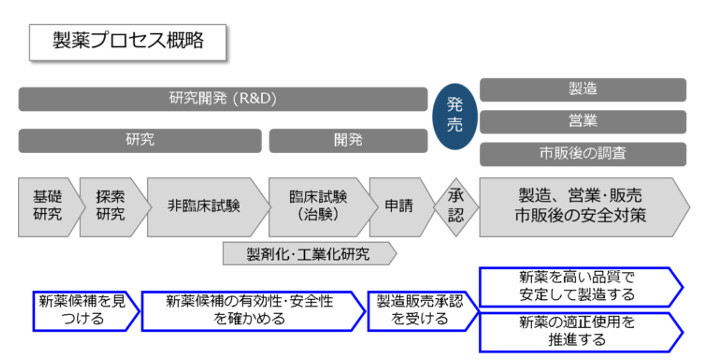
では、一つの新薬に対する製薬のプロセスを簡単に追ってみましょう。
医薬品は生命に関わるということで、厳しい法的規制を受けます。医薬品は製品ごとに製造販売の承認を厚生労働省より受ける必要があります。
製薬に関する業務は、製品が世に出るまでの研究開発と、承認後(市販開始後)の製造・販売、営業、市販後調査・安全対策に大別できます。
市販までの大まかな流れは次の通りで、この過程を「創薬」と呼びます。
承認を得た製品は医療現場で使用されますが、市販後にも製薬会社が実施すべきことがあります。
医薬品は研究開発の過程で様々な試験を実施しますが、承認を受けた段階においてわかっていることはごく一部であり、市販後に多くの患者に使用されてから得られる情報も多くあります。
このことから、市販後にも情報を蓄積し、医療現場でより良い形で使用してもらう活動が必要になります。
市販後に蓄積される情報によって、より有効・安全な医薬品に成長させることを「育薬」と呼びます。
医学・薬学の知見・データを基に、どういう病気・メカニズムにどういう効果を狙うのかという創薬の標的を定め、これに対して目的の効果を示す基本化合物(リード化合物)を探索します。
更により有効性・安全性・物性に優れた化合物を探索していくことで最適化を行い、医薬品の候補を選定します。
尚、目的とする効果を示す物質を評価して篩にかけることを「スクリーニング」、効果を示すかどうかを実験的に確認する評価方法(試験系)を「スクリーニング系」と呼びます。
候補化合物が決まったら、有効性・安全性・品質の面で薬としての条件を満たせるのか、証拠固めをしていきます。
いきなりヒトに投与するのは適当ではありませんので、まずは非臨床試験の段階として、医薬品の候補に対して動物等を用いて有効性・安全性を評価します。並行して品質面の試験も実施します。
非臨床試験において有効性・安全性の確認ができたら、ヒトを対象にした試験に移ります。この臨床試験では、医薬品の候補に対して段階的にヒトに投与することにより、有効性・安全性を評価していきます。
効果を示す薬効成分は、そのまま(薬効成分の原末)で医療に使われることはなく、実際に患者に使用できる形に加工(製剤化)します。薬剤の使用方法や物質の性質等を考慮して、最適な製剤を設計します。
また、安全で高品質な製品を実際の生産ラインで、安定的に(かつ安価に)生産するための研究(工業化研究)も行われます。
医薬品を発売するためには、承認を受ける必要があります。
研究開発の過程で得られた研究開発の成果を申請資料として纏め、製造販売の承認取得のための申請をします。承認権限は厚生労働省にありますが、審査の実務は独立行政法人医薬品医療機器総合機構(PMDA)が担います。
審査の過程では、申請内容に関して当局とのやりとりがあります。追加のデータの提出を求められることもありますし、場合によっては追加の試験が必要になることもあります。
審査の結果として承認されると、薬価収載された後に発売となります。
発売後において、新薬を製品として高い品質で安定して製造する必要があります。
製造工程の主な流れは以下の通りです。
原料 ⇒ 原薬 ⇒ 製剤 ⇒ 包装 ⇒ 出荷
製造所では、医薬品の製造工程にミスが起きないようにしっかり管理し、製品の品質を守る責任があります。
また製造販売業者(製薬企業)は、医薬品の製造から出荷、市販後まで一貫した品質管理を実施する必要があり、そのために以下の責務を負います。
前述の通り、発売時点で得られている情報には限界があり、市販後にも情報を蓄積し、医療現場でより良い形で使用してもらう活動が必要となります。PMS(Post Marketing Surveillance)は、医薬品の製造販売後に実施される有効性・安全性の確保のための活動です。
発売直後は使用患者数が増加することから未知で重篤な副作用が見つかることがあり、特に安全面で留意が必要です。そのため、販売を開始した後の6ヶ月間は「市販直後調査」を実施します。これは、重篤な副作用等の発生を迅速に把握するとともに、安全性に関する情報を医療機関へ確実に提供することで適正使用を促すものです。
その後も、「副作用・感染症報告制度」として実際の医療の現場で発現した副作用と思われる症例を収集し、評価したうえで必要な安全対策を講じることが義務付けられています。また定期的にPMDAへ安全性に関する報告を実施します(「安全性定期報告」)。
また、医療上の有効性・安全性・品質等を改めて評価する制度として製造販売後調査があり、この活動を通じて、製造販売後の医薬品の適正な使用方法を確立していきます。
新医薬品(新薬)に対しては、承認後4-10年(通常8年、承認区分に基づく)の使用成績等に関する調査を実施することで、承認時の有効性・安全性を再確認するという、「再審査」制度があります。これにより、実際の医療現場における実績を踏まえて、医薬品の評価が確立されます。
医薬品を適切に使用するためには、製品に関する情報が重要です。
医薬品の基本情報を記述したものが添付文書です。加えて、医師・薬剤師向けにより詳細な解説書が発信されます。
製造販売を開始するにあたっては、研究開発の過程から得られた情報を医療現場に提供します。
製造販売後には、実際の医療現場で使用される中で情報の収集・評価が行われます。例えば、副作用が出た場合には安全性情報に関する緊急安全性情報・安全性速報が医療現場に届けられ、必要に応じて添付文書の改訂が行われます。このように、適正使用・安全対策のために医療現場に対して製品に関する情報が提供されます。
製薬のプロセスは、研究開発の過程だけでなく、製造販売後も含めて医薬品を構成する情報について有効性・安全性・品質に関する情報を積み重ねていくプロセスであるということができます。
私たちの健康に大きな役割を担う医薬品。
その萌芽・誕生から退役までのライフサイクルにおけるプロセスや情報は厳しく重要なものです。そうした製薬のプロセスや情報を支えるITを介して、日立医薬情報ソリューションズは人々の健康に貢献していきます。
2019年08月23日
吉田 亜登美